
Hemostatic Mechanisms and Metastasis (Developments in Oncology)
There could be mechanisms of cell resistance due to the modification of the endothelial receptor and induction of APC under the influence of tumor proinflammatory cytokines [ 39 , 40 ]. Protein S—cofactor PrC whose activity can be inhibited by circulating paraproteins [ 24 ]. Decreased levels of ATIII are found in patients with malignant diseases and decreased survival [ 41 , 42 ]. Overexpression of heparanase induces increase of functionally inactive TFPI in plasma 36 , a putative role of a tumor suppressor gene [ 43 , 44 ]. Tumor cells expressed on their cellular surface all proteins necessary for regulation of the fibrinolytic pathways.
Deregulation in generating normal fibrinolytic activity was observed in patients with solid tumors and is a mechanism for the development of thrombotic tendency [ 45 ]. Plasminogen activator inhibitor-1 PAI-1 —important regulator of plasminogen activity. PAI-1 is overexpressed in different types of tumors [ 21 ]. Genomic sequencing of hepatocytes expressing the MET oncogene demonstrated significantly increased expression of the gene for PAI-1 and COX-2 that corresponded with triple increase in levels of circulating plasma proteins [ 46 ].
Thrombin-activated fibrinolysis inhibitor TAFI —blocks the binding of plasminogen to fibrin. Reported elevated levels of TAFI in patients with cancer and thromboembolic events were compared to patients with acute venous thrombosis and normal controls [ 47 , 48 ]. Immunohistochemical studies demonstrate the expression of fibrinolytic inhibitors only in tumor cells, which could explain the absence of fibrinolytic activity in some tumors [ 50 ]. Blood vessels and endothelial cells play a major role in the control of the processes of hemostasis, thrombosis, and inflammation. Endothelial tromboregulation is accomplished by selective expression of mediators autacoids and cell adhesion molecules in response to specific agonists.
Intact endothelium is anticoagulant and profibrinolytic under physiologic conditions. Malignant process causes deregulation of endothelial homeostasis, which can be defined more precisely as activation rather than damage as endothelial cells alter their functional capacity and acquire new properties in the absence of violation of tissue integrity [ 51 ].
Factors of endothelial activation in the presence of malignant process include the following: Endothelial expression of selectins and ligands from the immunoglobulin superfamily is increased under the effect of tumor-induced cytokine and cellular interactions. Through their silencing by small interfering RNA processes such as endothelial proliferation, adhesion and capillary formation are influenced [ 54 ].
Thrombomodulin TM —important endothelial cell-associated receptor that acts as direct anticoagulant. Binding of thrombin activates PrC system and inactivates the proteolytic degradation of procoagulant substrates [ 55 ]. Circulating tumor cytokines decreases TM levels causing degradation of its molecule and increased endothelial expression of TF [ 56 ]. There is inverse proportional relationship between TM expression and cellular proliferation in vivo. Downregulated to absent TM expression is found in metastatic foci, while forced expression of TM in transgenic mouse models of squamous cell carcinoma lacking TM expression leads to a differentiated epithelial-like phenotype—effect regulated by Snai1 transcription factor [ 57 ].
Interference of the TM-PrC system that occurs as a result of lower TM and development of acquired resistance to PrC of cellular type is one of the factors for procoagulant conversion of endothelium. Cytokine-activated endothelium releases high-molecular complexes of vWF, which are hyper-reactive to platelet aggregation, thrombus formation, and adhesion, whereas expression of ADAMTS13 depolymerase is suppressed [ 58 ].
Local procoagulant activity of endothelium is potentiated further by the effects of heparanase, which induces expression of TF and simultaneously dissociates its inhibitor TFPI from the endothelial cell surface [ 59 ]. And last but not least, the endothelial cells as a primary source of fibrinolytic activators participate in the induction of hypofibrilinolitic state by defective secretion of plasminogen activators in enhancing the expression of a fibrinolytic inhibitor PAI-1 [ 60 ]. Different mechanisms of microvascular dysfunction in combination with activated coagulation are the main pathogenetic factors in the development of thrombotic microangiopathy in malignancies.
Increased angiogenic activity is due to complex processes in which fully differentiated, non-proliferating endothelial cells acquire invasive, migratory, and proliferative properties. At the same time, many of these processes are coupled with regulatory coagulation processes.
For example, endothelial growth factor VEGF, secreted by the tumor cells, increases endothelial expression of TF, which causes inverse decrease in the expression of the negative angiogenic regulator—thrombospondin [ 62 ]. Synthesis and secretion of another potent proangiogenic cytokine—interleukin-8 IL-8 from endothelial cells—are increased, and the effect is dose dependent on the levels of fibrin deposits [ 63 ].
A similar mechanism is responsible for fibrin-induced expression of the gene for TF from umbilical vascular endothelial cells [ 64 ]. Additionally, thrombin and coagulation degradation products mediate haptotaxis of endothelial cells by selective exposure of a set of integrins. Thereby, they orientate the formation of capillaries and vasculogenesis in the direction of the angiogenic stimulus [ 65 , 66 ]. Tie2 is an endothelial-specific receptor tyrosine kinase whose ligands are angiopoietin-1 Ang-1 and angiopoietin-2 Ang Angiopoietin-1 is secreted by perivascular cells and in complex with Tie2 stabilizes the endothelium in quiescent state and potentiates the maturation of blood vessels.
It is overexpressed during tumor angiogenesis and is responsible for pro-angiogenic endothelial conversion [ 67 ]. Tumor blood vessels express structural and functional abnormalities such as abnormal vascular permeability and increased potential for rapid growth and remodeling due to overexpression of Ang-2 [ 68 ]. Experimental data support the interdependence of the processes of procoagulant and pro-angiogenic endothelial conversion.
Thrombin-induced angiogenesis in an in vivo model of chorion allantoic membrane is accompanied by a double increase in expression of mRNA, encoding VEGF and Ang2 [ 70 ]. Endothelium performs a crucial role as a regulatory nexus in the processes of hemostasis and angiogenesis. The complexity of interactions allows on the one hand multiple targeting of several pathological mechanisms involved in angiogenesis and tumor progression and on the other hand mediates expression of side effects associated with the system of hemostasis. In the context of Virchow triad, disturbed blood flow is a predisposing factor for thrombosis.
Venous stasis is most often secondary—due to external compression of local or metastatic tumor masses, adenopathy—inflammation caused by the tumor. The main mechanism involves inadequate clearance of coagulation factors and local endothelial hypoxia that induces endothelial expression of TF and platelet activating factor, increased leukocyte adhesion and platelet activation. Additional predisposing factor is the prolonged immobilization of cancer patients both in hospital and at home. Additive effect of disturbances clearance of activated coagulation factors and hypoxic endothelial damage in conditions of prolonged immobilization contribute to the development of a thrombotic process.
Besides its major role in hemostasis, TF has been identified as an important signaling receptor in cancer biology. Ample preclinical evidence has accumulated over the past decade, implicating TF as an important effector in the processes of tumor initiation, growth, angiogenesis, and metastasis. Moreover, this has led to the development of approaches exploring TF as a potential target for anticancer therapy [ 71 ]. TF is overexpressed in many types of human tumors, including breast cancer, pancreatic cancer, gastric cancer, prostate cancer, colorectal cancer, non-small-cell lung cancer, melanoma, leukemia, lymphoma, esophageal cancer, hepatocellular carcinoma, brain glioblastoma, but not in their normal tissue counterparts [ 1 ].
TF overexpression in cancer cells has been correlated with tumor progression and unfavorable prognostic indicators such as increased angiogenesis, advanced disease stage, and resistant phenotype [ 72 — 74 ]. Therefore, TF overexpression in situ could be considered a biomarker for solid tumors. Enhanced TF expression in cancer has been reported to be an oncogenic-driven event. In colorectal cancer, activation of the K-ras oncogene and loss-of-function mutation of p53 result in constitutive activation of mitogen-activated protein kinase MAPK and PI3K pathways leading to increased TF expression [ 75 ].
In medulloblastoma cell lines, TF expression has been shown to result from mutation in the c-Met oncogene and subsequent activation of Src kinases [ 77 ]. It has been observed that a certain subset of tumor cells, known as cancer stem cells, which constitutively express activated oncogenes and are capable of undergoing multilineage differentiation, are characterized by TF abundant phenotype [ 5 ]. Moreover, enhanced TF expression is observed during the processes of epithelial-to-mesenchymal-transition, whereby epithelial cells acquire a mesenchymal, more aggressive and motile phenotype [ 78 ].
This indicates that TF is possibly involved in maintaining cancer cell self-perpetuance. There is a structure function dependency in TF mode of action. TF plays a role in cancer progression both by initiating tumor growth and by promoting efficient tumor cell dissemination. Tumor-promoting activities of TF occur via non-hemostatic mechanism and can be attributed to the cytoplasmic domain signaling dependent mostly on the activation of the protease activated receptor 2 PAR2. Prometastatic properties of TF can rather be coupled with its extracellular domain and the subsequent generation of thrombin, which, as a potent growth factor, exhibits further pleiotropic cellular effects.
TF-mediated signaling is critical for both physiological and pathological angiogenesis. TF deficiency in murine knockout experiments caused early embryonic lethality due to impaired vasculature development [ 79 ]. It has been revealed that involvement of TF cytoplasmic domain in several transduction cascades accounts for the production by tumor cells of angiogenic cytokines and contributes to increased angiogenesis in a paracrine fashion [ 81 ]. In addition to PAR2 signaling, the cytoplasmic domain of TF can be phosphorylated independently of f. The relationship between TF and VEGF has been extensively studied and is manifested by reciprocal co-stimulation of their expression profiles.
Experimental ex vivo and in vivo studies have further supported the TF-VEGF interrelationship by finding increased co-expression on tumor sections and their association with increased angiogenesis and malignant potential in human tumors [ 73 — 75 , 88 , 89 ]. This is evidenced by studies on cancer cell lines, where overexpression of TF by cancer cells conferred growth advantage compared to cell lines expressing low levels of TF [ 71 , 86 ].
Studies on selective targeting of different domains of the complex TF: Prometastatic properties of TF can be attributed to its extracellular domain, which is required for its major role in triggering coagulation. The extracellular mutant domain TFmut has markedly diminished function for activation of f. X, while full-length or cytoplasmic tail-deleted TF retains its procoagulant activity [ 85 , 92 ].
Tumor cells become encapsulated in fibrin and platelet-rich thrombi, being protected from the host immune defense and arrest in the microcirculation. Local thrombin formation facilitates arrest of tumor cells to the vessel wall by upregulation of adhesion molecules and strengthening cell-to-cell junctions [ 93 ]. Fibrin matrix in the tumor stroma builds itself a multifunctional scaffold rich on growth factors such as platelet derived growth factor PDGF , transforming growth factor TGF , fibroblast growth factor FGF , that is not only protective, but also promotes matrix-cell interactions necessary for neovascularization [ 94 ].
Thrombin has also an important function in angiogenesis by inducing the activation of endothelial-secreted collagenase type IV, which degrades basement matrix proteins and collagen during neoangiogenesis [ 98 ].
Hemostatic Mechanisms and Metastasis : Kenneth V. Honn :
In vivo experimental models of metastasis demonstrated dramatic increase of lung metastasis with thrombin-treated tumor cells compared with untreated cells [ 99 ]. Taking into account the specific biologic role of TF in the malignant tissue, detecting circulating TF in cancer patients might be informative of active disease and ongoing processes of matrix reorganization, cell destruction, and neovascularization. The physiologic role of fibrinolysis is dissolution of the fibrin clot and collagen degradation exerted by the action of plasmin.
Generation of plasmin, the main enzyme in fibrinolysis, occurs upon activation of plasminogen by the tissue plasminogen activator tPA and the urokinase plasminogen activator uPA. Therefore, function of the urokinase plasminogen activator uPA and its high-affinity cellular receptor uPAR is critical for fibrinolytic activities including targeted degradation of the basement matrix.
Moreover, uPAR is motile within the cellular membrane, which allows its allocation at the cellular front of desired direction for proteolysis [ ]. Under normal conditions, the process of active proteolysis is tightly controlled by the proteolytic systems. Extracellular matrix proteolysis acts at all stages of the metastatic cascade: The cellular receptor for the urokinase plasminogen activator uPAR is a key molecule for efficient pericellular proteolysis.
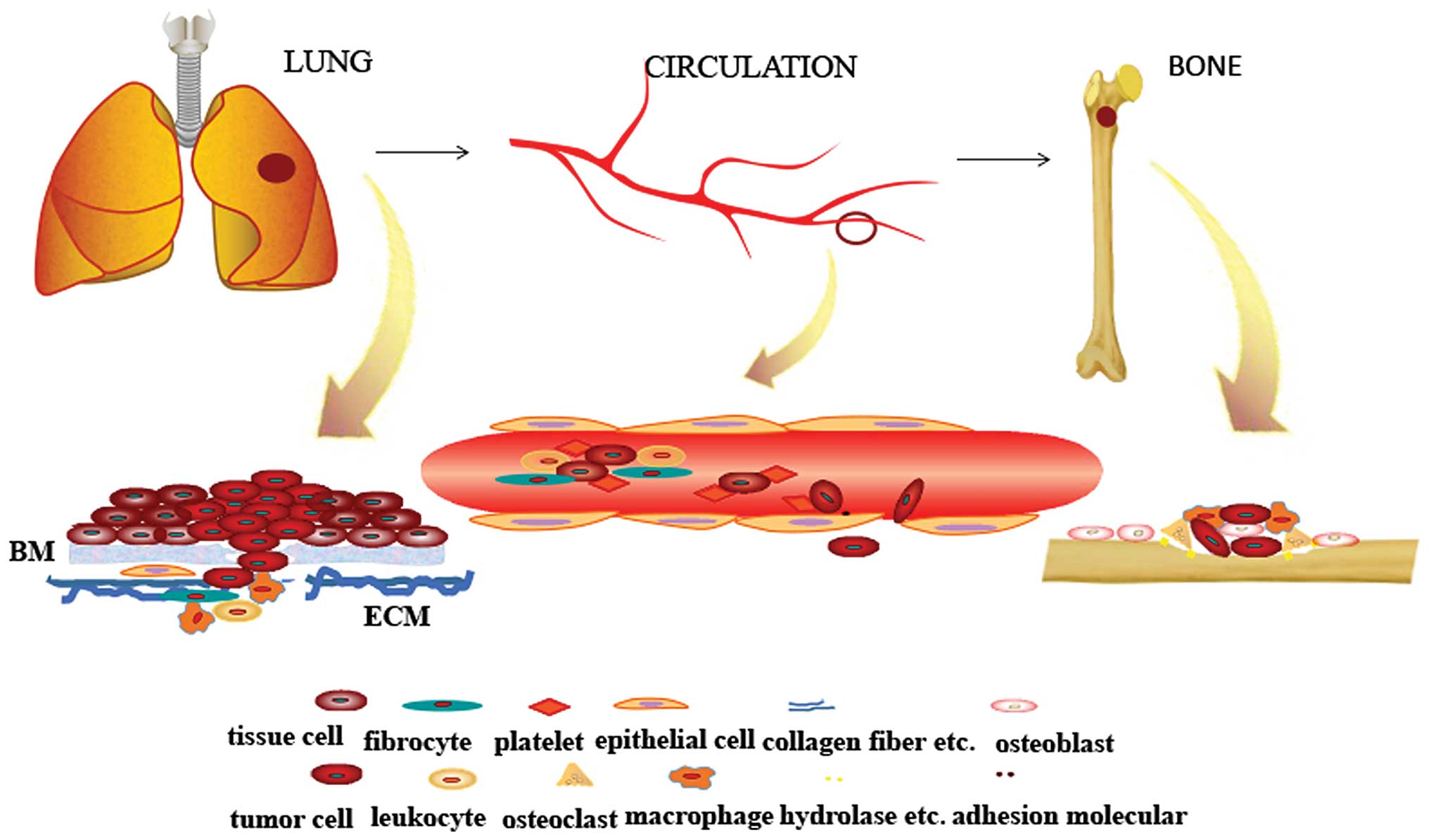
Hemostatic System in Malignancy: Providing the “Soil” in Metastatic Niche Formation
It is associated with advanced disease and is independent adverse prognostic factor for survival [ ]. Direct involvement of uPAR in processes of tumor biology characterizes it as a hallmark of the malignant invasive phenotype. Invasive potential of tumor cells in chorion-allantois membrane of chicken embryos correlates with uPAR-associated proteolytic activity [ ]. Expression of uPAR gene by tumor cells is required for vascular intravasation, whereas uPAR gene expression decreases invasive potential of transformed fibroblasts in vitro [ ].
Experiments with anti-uPAR inhibitory antibodies demonstrate reduction of the matric proteolytic activity [ , ]. Levels and activity of uPAR are regulated at the transcriptional level by oncogene-controlled promoter activation. AP2 binding motif is required for constitutive overexpression of uPAR promoter activity in invasive tumor cells after stimulation by the tumor promoter phorbol acetate. Downregulation of promoter activity as in deletion of the AP1 binding activity has been observed in tumor clones with K-ras allelic deletion. The knockout effect is accompanied by significant reduction of uPAR expression and tumor-associated proteolysis [ ].
Elevated uPAR expression is due to transcriptional activation secondary to increased binding of SP1 to the complementary promoter motif. Fibrinolytic system components can be identified as determinants of invasion in tumor biology and reflect the metastatic potential of tumors. The Formation of Metastases from Micrometastases. Overview on Blood Coagulation Proteins.
Structural Characteristics of Serine Proteinases. The Role of Vitamin K. Regulation of Blood Coagulation. The Relationship between the Intrinsic and Extrinsic Pathways. Inhibitors of Blood Coagulation. Role of Platelets in Blood Coagulation. The Microinjury Hypothesis and Metastasis. The Silent Sector of the Metastatic Cascade. Hemostatic Abnormalities in Tumor-Bearing Animals. Animal Models of Dissemination. Evidence for a Tumor Proteinase in Blood Coagulation. Purification of Cancer Procoagulant. Characterization of Cancer Procoagulant.
Distribution of Cancer Procoagulant. Tissue Factor as the Tumor Procoagulant. Rationale For Cancer Procoagulant. Role of Procoagulants in Metastasis. Implications for Tumor Growth and Metastasis. Introduction and Historical Background. The Nature of Fibrin Deposits in Tissues. Fibrin is a Component of the Tumor Microenvironment. Guinea Pig Hepato bile duct Carcinomas. Infiltrating ductal breast carcinoma.
Hodgkin's disease and other malignant lymphomas. Factor X cleaving activities. Shed plasma membrane vesicles. Thrombin-like and factor XIII-like activities. Perspectives on the Role of Platelets in Hemostasis and Thrombosis. Coagulant Activities of Platelets. Role of Platelets in Contact Activation. Interaction of Platelets with Coagulation Factors.
The Interaction of Platelets with Factor V. Spontaneous shedding of vesicles by A. Activity of vesicles isolated by sucrose density gradient centrifugation. Isotonic low ionic strength medium. Tumors which Aggregate Platelets via the Generation of Thrombin. Tumor Proteolytic Enzymes and Metastasis. Tumor Cysteine Proteinases and Metastasis. Cysteine Proteinase Activity in Tumors. Cysteine Proteinase Activity Released from Tumors. Tumor Cysteine Proteinases and Platelet Aggregation. Arachidonate Metabolism in Platelets and Blood Vessels. Metabolism of Prostaglandins and Thromboxanes.
Bestselling Series
Platelets, Coagulation and Metastasis. Prostacyclin, Thromboxanes, Platelets And Metastasis. Prostacyclin effects on platelet-induced tumor cell adhesion in vitro. Prostacyclin effects on metastasis in vivo. Role of endogenous PGI2 in tumor metastasis.
Hemostatic Mechanisms and Metastasis
Effect of agents that stimulate or synergize with endogenous PGI2 production. Agents that alter PGI2 metabolism. Normal Platelet and Vessel Wall Activity. Platelets and Tumor Growth. Production of Prostaglandins by Tumor Cells. Role of Prostaglandins in Tumor Metastasis. Role of Platelets in Tumor Metastasis.
Antiplatelet Effects of Calcium Channel Blockers. Platelet Aggregation Induced by Chemical Agonists.
- chapter and author info.
- Top Authors.
- !
- .
- Gravesend Through Time.
- Home Front.
Platelet Aggregation Induced by Tumor Cells. Antmetastatic Effects of Calcium Channel Blockers. Evidence for the Antimetastatic Effects of Coumarin Derivatives. Anticoagulants and Tumor Metastases.